今天给同学们分享一篇生信文章“Machine learning algorithms reveal potential miRNAs biomarkers in gastric cancer”,这篇文章发表在Sci Rep期刊上,影响因子为4.6。

结果解读:
数据来源本研究包括348名男性(占64.9%)和188名女性(占35.1%)的临床病理信息,这些信息从TCGA下载并总结。平均年龄为65.3岁,约有250人(占46.6%)患有晚期胃癌。
相关性分析在临床病理数据中,只有疾病阶段与癌症显著相关。衡量相关性的显著标准被认为是 p 值小于 0.05(图2A)。

如材料和方法部分所述,临床信息和测序的原始数据来源于TCGA数据库。根据所述标准,选择了536个样本进行进一步研究,其中约465个与GC患者相关,72个与年龄和性别匹配的对照组相关。
数据预处理和不同表达的miRNA(DeMiRs)的鉴定
数据集包括1882个miRNA,在使用Limma软件包和R软件进行归一化后,减少到220个miRNA。在使用热图进行处理的步骤中,选择了最重要的特征(图2B),并使用机器学习算法进行分类。然后,使用五种算法(SVM、dts、rf、逻辑回归和knn)以及四种不同的指标(准确率、f1得分、ROC曲线和混淆矩阵)进行检验,最后根据这四个指标得到的分数,选择了SVM算法作为最准确的算法(DTS,准确率:88%,AUC = 47%;随机森林,准确率:93%,AUC = 39.5%;SVM,准确率:93%,AUC = 88.5%;KNN,准确率:93%,AUC = 41.7%;逻辑回归,准确率:93%,AUC = 88%)。混淆矩阵也可以在图2C中看到。因此,在胃癌中选择了一个包含29个miRNA的列表,其中有5个显著上调表达和24个显著下调表达,以进行进一步分析(图3)。

ROC曲线分析的结果显示了hsa-miR-29c的诊断能力(AUC为0.7,敏感性为0.5,特异性为0.8,截断值为0.88),当与hsa-miR-93结合时,其诊断能力得到改善(组合的AUC为0.76,敏感性为0.69,特异性为0.73,截断值为0.86)(图2D)。
demiRs的生存分析
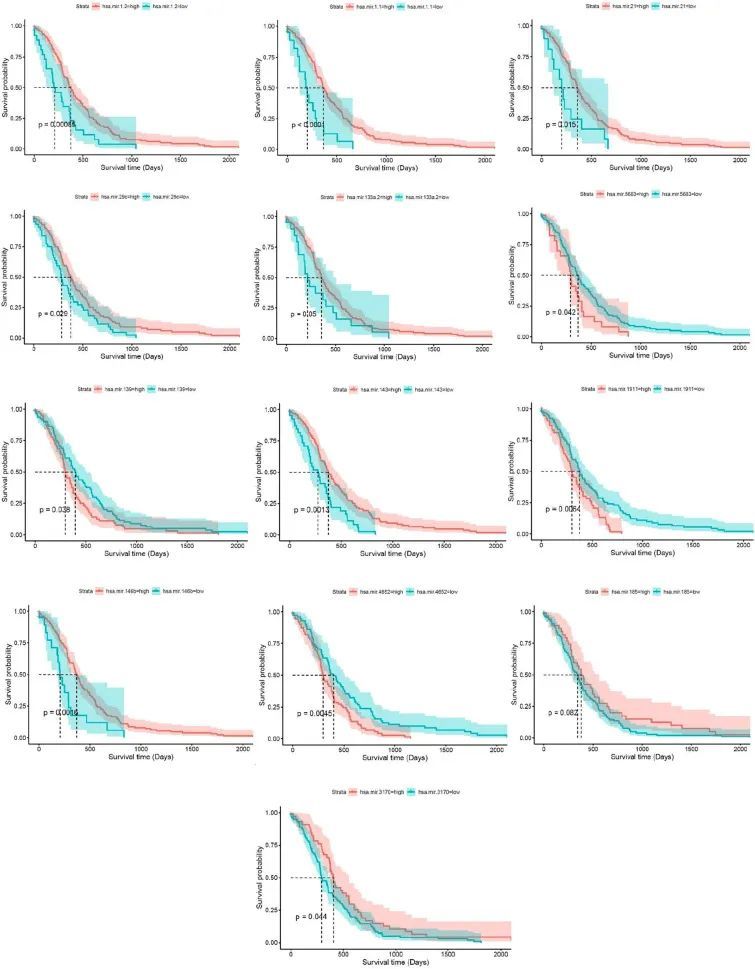
使用SPSS 进行了demiR的生存分析,p值被认为是<0.05。结果表明,有13个miRNA(Hsa-miR-21、Hsa-miR-146b、Hsa-miR-185、Hsa-miR-1.1、Hsa-miR-1.2、Hsa-miR-143、Hsa-miR-4652、Hsa-miR-1911、Hsa-miR-29c、Hsa-miR-3170、Hsa-miR-139、Hsa-miR-5683和Hsa-miR-133a.2)具有预后能力(图4)。
在材料和方法部分提到的在线网络服务器中,使用机器学习算法得出的29个候选microRNA中,hsa-miR-21、hsa-miR-133a、hsa-miR-146b、hsa-miR-29c和hsa-miR-204的表达水平在(EXP00118(GSE28700)、EXP00131(GSE23739)、EXP00230(GSE26595)、EXP00268、EXP00270(GSE54397)、EXP00326(GSE31568)、EXP00337(GSE59856)、EXP00404、EXP00460(GSE93415)、EXP00524(GSE106817)、EXP00405、EXP00118(GSE28700)、EXP00406、EXP00666、EXP00444(GSE78775)、EXP00476(GSE99415)、EXP00316(GSE77380)和EXP00175(GSE33743)中得到了高度验证,使用miRPathDB在线服务器进行了热图分析(https://mpd.bioinf.uni-sb.de/,图5A)。

miRNA靶向预测是通过使用几个数据库完成的,包括:miRwalk、miRdb和Targetscan。使用一个在线Venn图数据库,确定了包含407个共同基因的列表(图5B)。
蛋白质相互作用网络分析在前一步中预测的候选基因被提交到STRING数据库,根据材料和方法中提到的标准构建了一个蛋白质相互作用网络(PPI网络)。为了获得具有重要作用的关键基因,PPI网络随后被导入并通过Cytoscape软件进行可视化。使用Cytohubba工具选择了基于度数的100个得分最高的基因(图5C)。最后,这些关键基因被导入到STRING数据库中重新分析PPI网络(图5D)。
功能分析为了揭示所选关键基因的作用,使用R软件进行了富集分析。结果表明,就分子功能而言,关键基因主要富集于转录因子结合、酶结合、RNA聚合酶II顺式调控区域特异性DNA结合、蛋白质结合、双链DNA结合、阻滞素家族蛋白结合、特异性DNA结合和染色质结合。在生物过程方面,大多数基因富集于miRNA介导的翻译抑制、宿主对病毒转录的正调控、基因表达的遗传印记调控、miRNA介导的基因沉默产生的miRNA、Wnt信号通路、钙调节通路、细胞衰老调控、基因表达的负调控和表观遗传基因沉默。染色质、真染色质、核质、非膜结合细胞器和细胞质是富集程度最高的细胞组分。使用KEGG数据库识别重要的信号通路表明,候选关键基因主要涉及到胶质瘤、黑色素瘤、前列腺癌、非小细胞肺癌、肾细胞癌、GnRH分泌、醛固酮调节的钠重吸收以及胰腺癌(图6)。

总结
总之,使用机器学习算法鉴定了29种miRNA,随后的分析显示,一组四种miRNA具有很高的诊断和预后能力,包括hsa-miR-21、hsa-miR-133a、hsa-iR-146b和hsa-miR-29c,这已通过多个数据集进行了验证。这项研究强调了机器学习作为预测癌症生物标志物的替代选择的重要性。然而,为了进一步验证,建议在实验室的几个队列中分析候选miRNA。
